![]()
Matilda is a multi-task framework for learning from single-cell multimodal omics data. Matilda leverages the information from the multi-modality of such data and trains a neural network model to simultaneously learn multiple tasks including data simulation, dimension reduction, visualization, classification, and feature selection.
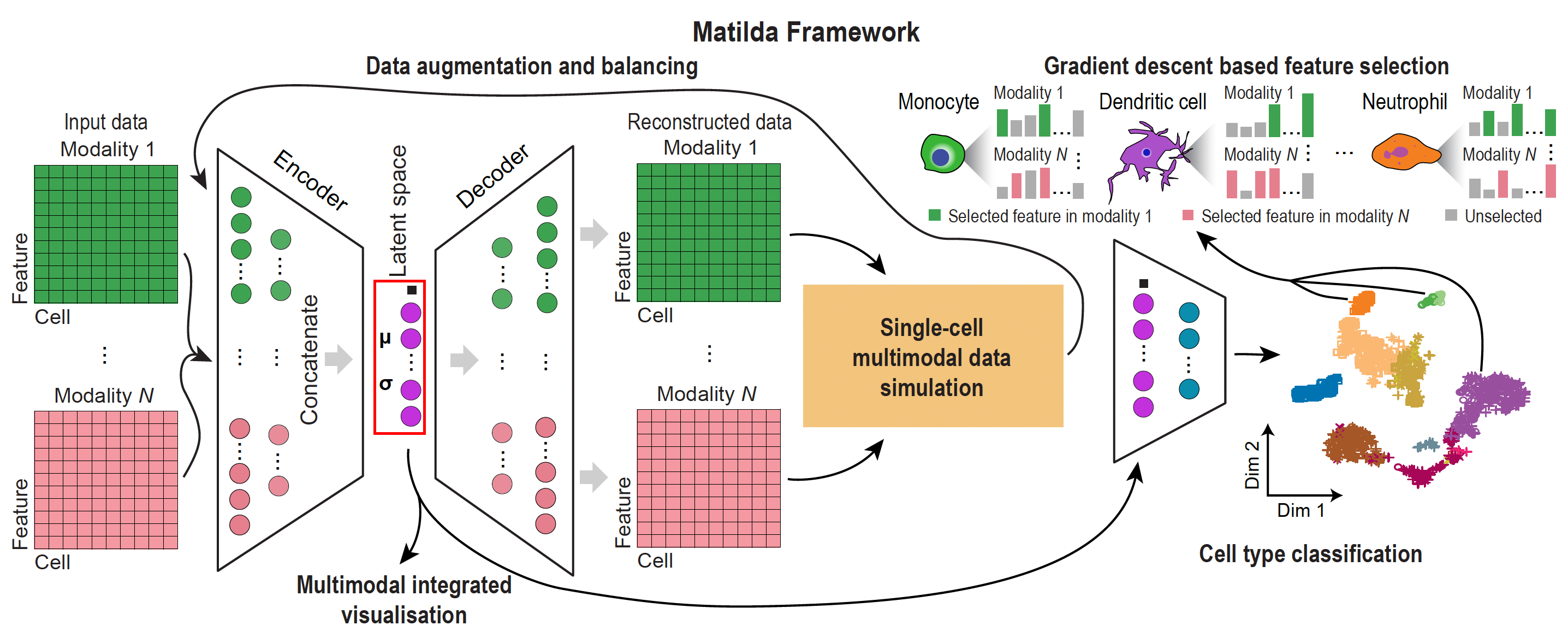
For more details, please check out our publication.
This repository packages Matilda as a Python package (pip install matilda-sc, import matilda) and an R package, wrapping the unchanged published engine, with documentation and runnable Colab tutorials.
- Documentation: https://pyanglab.github.io/Matilda/
- Python tutorial (Colab):
- R tutorial (Colab):
- R package: https://github.com/PYangLab/Matilda/tree/main/matilda-r (SingleCellExperiment-based, Bioconductor-style)
Python (import name matilda; PyPI distribution matilda-sc):
pip install "git+https://github.com/PYangLab/Matilda.git" # once on PyPI: pip install matilda-scR (Python is provisioned automatically by basilisk, so you never install or manage it):
remotes::install_github("PYangLab/Matilda", subdir = "matilda-r")Matilda ships as a Python package (import matilda) and an R package (matilda, operating on a
SingleCellExperiment). Both call the same engine, so on the same hardware they give the same
result. Pick the interface that matches your workflow; see the documentation
for both APIs and full tutorials.
Work with in-memory AnnData (or arrays, or file paths) and get results back as objects. After
train, there is one verb per task: classify / reduce / markers / simulate.
import matilda
# rna/adt/atac: AnnData | ndarray | scipy.sparse | path | None
# labels: a vector, an `.obs` column name, or a .csv path (string or numeric labels)
fit = matilda.train(rna, adt=adt, atac=atac, labels="cell_type")
res = matilda.classify({"rna": q_rna, "adt": q_adt, "atac": q_atac},
model=fit, query_labels=q_labels)
res.predictions # DataFrame: cell_id, real, predicted, probability
res.celltype_accuracy # DataFrame: celltype, accuracy, n
lat = matilda.reduce({"rna": rna, "adt": adt, "atac": atac}, model=fit) # lat.latent
mk = matilda.markers({"rna": rna, "adt": adt, "atac": atac}, model=fit, labels="cell_type") # mk.markers
sim = matilda.simulate({"rna": rna, "adt": adt, "atac": atac}, model=fit, celltype="B.Naive", n=200, labels="cell_type") # sim.simulatedThe modality combination is inferred automatically (RNA only, RNA+ADT for CITE-seq, RNA+ATAC for
SHARE-seq, all three for TEA-seq). classify reconciles features automatically: if the query is
missing some of the model's features it takes the per-modality reference intersection (real values,
no zero-padding), retrains, and classifies; res.retrained / res.common_features report what
happened. See the Quickstart and
API reference for the full surface.
If you use Matilda, please cite the Matilda paper (see the Citation page).